Sequence analysis in bioinformatics refers to the process of analyzing biological sequences, such as DNA, RNA, or protein sequences, to extract meaningful information and uncover patterns, structures, functions, or evolutionary relationships within these sequences. It involves a range of computational methods, algorithms, and tools to interpret and make sense of the vast amount of sequence data generated through various experimental techniques.

Sequence analysis
Sequence analysis encompasses several key aspects:
- Sequence Alignment: Aligning sequences to identify similarities, differences, conserved regions, or evolutionary relationships. Tools like BLAST and Clustal Omega are used for this purpose.
- Annotation and Functional Prediction: Identifying genes, regulatory elements, coding regions, motifs, or functional domains within sequences. Predicting protein functions based on sequence similarities or structure predictions.
- Homology Search: Searching for sequences similar to a query sequence in databases to infer evolutionary relationships, identify orthologs or paralogs, or predict protein structures and functions.
- Motif and Pattern Discovery: Identifying conserved motifs, patterns, or regulatory elements within sequences using computational methods like MEME or HMMER.
- Phylogenetic Analysis: Constructing evolutionary trees or phylogenetic relationships between different sequences or organisms based on sequence data.
- Structural Analysis: Predicting or analyzing the 3D structure of proteins or RNA molecules based on their sequences using tools like Phyre2 or RNAfold.
- Comparative Genomics: Comparing genomes or genetic sequences across different species to understand evolutionary changes, gene families, or regulatory elements.
Sequence analysis is fundamental in various areas of biology and bioinformatics, including genomics, transcriptomics, proteomics, evolutionary biology, drug discovery, and understanding genetic diseases. It aids in unraveling the complexities of biological systems, identifying functional elements, and providing insights into the structure, function, and evolution of genes and proteins.
Following is an example for homology search done in Python:
from Bio.Blast import NCBIWWW, NCBIXML
# Perform a BLAST search (blastp) with a protein sequence against the NCBI nr database
query_sequence = "MTEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAAKKADRLAAEG" # Replace with your protein sequence
result_handle = NCBIWWW.qblast("blastp", "nr", query_sequence)
# Parse and process the BLAST results
blast_records = NCBIXML.parse(result_handle)
for record in blast_records:
print(f"Query: {record.query[:50]}...") # Display the first 50 characters of the query sequence
for alignment in record.alignments[:3]: # Display the top 3 alignments
print(f"Subject: {alignment.title[:50]}...") # Display the first 50 characters of the subject title
for hsp in alignment.hsps:
print(f"Length: {alignment.length}, E-value: {hsp.expect}, Identity: {hsp.identities}")
print(f"Alignment: {hsp.align_length} matching positions")
print(hsp.query[:50]) # Display the first 50 characters of the query alignment
print(hsp.match[:50]) # Display the first 50 characters of the match alignment
print(hsp.sbjct[:50]) # Display the first 50 characters of the subject alignment
print()Will output:
Query: unnamed protein product...
Subject: gb|EBF5727231.1| translation elongation factor Ts ...
Length: 130, E-value: 5.28488e-33, Identity: 59
Alignment: 60 matching positions
MTEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
M EITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
MAEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
Subject: gb|ECP7634291.1| translation elongation factor Ts ...
Length: 120, E-value: 6.51687e-33, Identity: 59
Alignment: 60 matching positions
MTEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
M EITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
MAEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
Subject: ref|WP_079741764.1| translation elongation factor ...
Length: 124, E-value: 6.71684e-33, Identity: 59
Alignment: 60 matching positions
MTEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
M EITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAA
MAEITAAMVKELRESTGAGMMDCKNALSETNGDFDKAVQLLREKGLGKAAThis code snippet uses Biopython’s NCBIWWW.qblast function to perform a protein BLAST search (blastp) against the NCBI non-redundant (nr) database. It then parses and processes the results, displaying details of the top three alignments found in the search.
Remember, BLAST searches can take some time, especially for larger sequences or databases, and require an internet connection to access the NCBI databases. Adjustments can be made to handle larger result sets or customize the output based on your specific requirements.
Sequence alignment methods
Sequence alignment methods are fundamental in bioinformatics, aiding in comparing and analyzing biological sequences such as DNA, RNA, or proteins. These methods aim to identify similarities, differences, and evolutionary relationships between sequences. There are two primary types of sequence alignment methods: pairwise alignment and multiple sequence alignment.
Pairwise Sequence Alignment:
- Objective: Aligns two sequences to identify regions of similarity or homology.
- Usage: Commonly used when comparing two sequences to find similarities, such as between a newly sequenced gene and a known reference.
- Algorithms: Dynamic programming algorithms like Needleman-Wunsch (global alignment) and Smith-Waterman (local alignment) are often used.
- Output: Provides an alignment highlighting matched (identical or similar) and mismatched positions between the two sequences.
Multiple Sequence Alignment (MSA):
- Objective: Aligns three or more sequences simultaneously to identify conserved regions, insertions, deletions, and evolutionary relationships.
- Usage: Useful for studying gene families, identifying conserved motifs, or understanding evolutionary patterns among related sequences.
- Algorithms: Methods like progressive alignment (e.g., Clustal Omega, MAFFT) or iterative refinement (e.g., T-Coffee) are employed to align multiple sequences.
- Output: Generates a multiple alignment showing conserved regions, gaps (insertions or deletions), and consensus sequences across multiple input sequences.
Both pairwise and multiple sequence alignment methods use scoring systems (such as substitution matrices like BLOSUM or PAM matrices) to assign scores to matches, mismatches, gaps, and extensions. These scores influence the alignment results by determining the optimal alignment configuration.
Pairwise alignment is more straightforward and computationally less intensive, making it suitable for comparing two sequences. Multiple sequence alignment is more complex due to the additional sequences involved and the challenge of aligning multiple sequences while considering their similarities and differences.
Overall, sequence alignment methods are crucial in bioinformatics for understanding sequence conservation, functional domains, evolutionary relationships, and structural or functional implications of sequence variations across different biological sequences.
This is an implementation of the Smith-Waterman (local alignment) algorithm in Python:
def smith_waterman(sequence1, sequence2, match=2, mismatch=-1, gap_penalty=-1):
# Initialize matrix to store scores
rows = len(sequence1) + 1
cols = len(sequence2) + 1
score_matrix = [[0 for _ in range(cols)] for _ in range(rows)]
# Fill the matrix with scores
max_score = 0
max_pos = (0, 0)
for i in range(1, rows):
for j in range(1, cols):
if sequence1[i - 1] == sequence2[j - 1]:
diagonal_score = score_matrix[i - 1][j - 1] + match
else:
diagonal_score = score_matrix[i - 1][j - 1] + mismatch
up_score = score_matrix[i - 1][j] + gap_penalty
left_score = score_matrix[i][j - 1] + gap_penalty
score_matrix[i][j] = max(0, diagonal_score, up_score, left_score)
if score_matrix[i][j] >= max_score:
max_score = score_matrix[i][j]
max_pos = (i, j)
# Traceback to find the aligned sequences
aligned_seq1 = ''
aligned_seq2 = ''
i, j = max_pos
while score_matrix[i][j] != 0:
if score_matrix[i][j] == score_matrix[i - 1][j - 1] + (match if sequence1[i - 1] == sequence2[j - 1] else mismatch):
aligned_seq1 = sequence1[i - 1] + aligned_seq1
aligned_seq2 = sequence2[j - 1] + aligned_seq2
i -= 1
j -= 1
elif score_matrix[i][j] == score_matrix[i - 1][j] + gap_penalty:
aligned_seq1 = sequence1[i - 1] + aligned_seq1
aligned_seq2 = '-' + aligned_seq2
i -= 1
else:
aligned_seq1 = '-' + aligned_seq1
aligned_seq2 = sequence2[j - 1] + aligned_seq2
j -= 1
return max_score, aligned_seq1, aligned_seq2
# Example usage
seq1 = "AGCACACA"
seq2 = "ACACACTA"
score, aligned_seq1, aligned_seq2 = smith_waterman(seq1, seq2)
print(f"Sequence 1: {aligned_seq1}")
print(f"Sequence 2: {aligned_seq2}")
print(f"Alignment Score: {score}")Will output:
Sequence 1: AGCACAC-A
Sequence 2: A-CACACTA
Alignment Score: 12Sequence similarity and homology
Sequence similarity and homology are fundamental concepts in bioinformatics, particularly in the context of molecular biology and genetics, relating to the comparison of biological sequences like DNA, RNA, or protein sequences.
Sequence Similarity:
Sequence similarity refers to the degree of likeness or resemblance between two sequences. It measures how much two sequences are alike in terms of their composition, structure, or function. Similarity can be assessed by aligning sequences and identifying regions where the nucleotides or amino acids match or have similar properties.
For example, in DNA sequences, similarity might involve matching identical nucleotides (A, T, C, G) or identifying regions where mutations or substitutions have occurred, leading to slight variations. In protein sequences, similarity could mean identifying regions with similar amino acids based on properties like charge, size, or chemical characteristics.
Homology:
Homology, on the other hand, implies a deeper evolutionary relationship between sequences. It indicates a common ancestry or origin for the sequences being compared. Sequences are considered homologous if they share a common evolutionary ancestor and have diverged over time due to mutations and evolutionary processes.
Homologous sequences might retain similar functions or structures despite undergoing changes over evolutionary periods. Homology can be classified into:
- Orthologs: Sequences in different species that evolved from a common ancestral sequence due to speciation. Orthologs often retain similar functions.
- Paralogs: Sequences within the same species that have evolved from a gene duplication event. Paralogs might have similar or divergent functions due to subsequent evolutionary changes.
Relation Between Similarity and Homology:
High sequence similarity often suggests the possibility of homology, but it doesn’t guarantee it. Similar sequences might arise due to convergent evolution or functional constraints rather than common ancestry. Therefore, sequence similarity is used as an initial indication of potential homology, and further analysis, like phylogenetic studies or structural comparisons, helps determine homologous relationships.
In summary, sequence similarity assesses the degree of likeness between sequences, while homology implies a shared evolutionary history between sequences, indicating common ancestry and potential functional or structural relationships.
Phylogenetic analysis and tree-building methods
Phylogenetic analysis is a process used in biology to study the evolutionary relationships between different organisms, genes, or species. It aims to reconstruct the evolutionary history or phylogeny, often represented as a phylogenetic tree or cladogram, which depicts the branching patterns and relationships among these entities.
Tree-building Methods:
- Distance-Based Methods: These methods estimate evolutionary distances between sequences or taxa based on genetic differences. Common distance metrics include Jukes-Cantor, Kimura, or Hamming distances. Algorithms like Neighbor-Joining and UPGMA (Unweighted Pair Group Method with Arithmetic Mean) use these distances to construct trees.
- Character-Based Methods: Also known as maximum parsimony, these methods analyze the characters (nucleotides, amino acids) at different positions in sequences. They seek the tree that requires the fewest evolutionary changes to explain observed differences among sequences.
- Likelihood-Based Methods: These methods use statistical models to estimate the probability of observed data given a particular tree topology and model of sequence evolution. Maximum Likelihood and Bayesian Inference are common likelihood-based approaches.
- Concatenation and Supermatrix Methods: These involve combining multiple genes or genomic regions into a single dataset for tree construction. This approach is particularly useful when analyzing large-scale genomic data.
Phylogenetic Analysis Process:
- Sequence Acquisition: Obtain sequences (DNA, RNA, or protein) from different organisms or genes of interest.
- Sequence Alignment: Align the sequences to identify homologous positions and understand their similarities and differences.
- Evolutionary Model Selection: Choose an appropriate evolutionary model that best fits the dataset based on the sequence characteristics (nucleotide or amino acid substitution rates, etc.).
- Tree Construction: Apply a tree-building method based on the chosen model to infer the evolutionary relationships among sequences. This involves generating a tree that optimally explains the observed similarities or differences.
- Tree Evaluation and Interpretation: Assess the robustness of the tree by statistical methods or bootstrapping to understand the confidence in inferred relationships. Interpret the tree to draw conclusions about evolutionary history, divergence times, and relationships among the entities.
Phylogenetic trees serve as powerful tools for understanding evolutionary relationships, aiding in various fields like evolutionary biology, systematics, epidemiology, and conservation biology. They provide insights into the evolutionary processes, common ancestors, and the diversification of life forms on Earth.
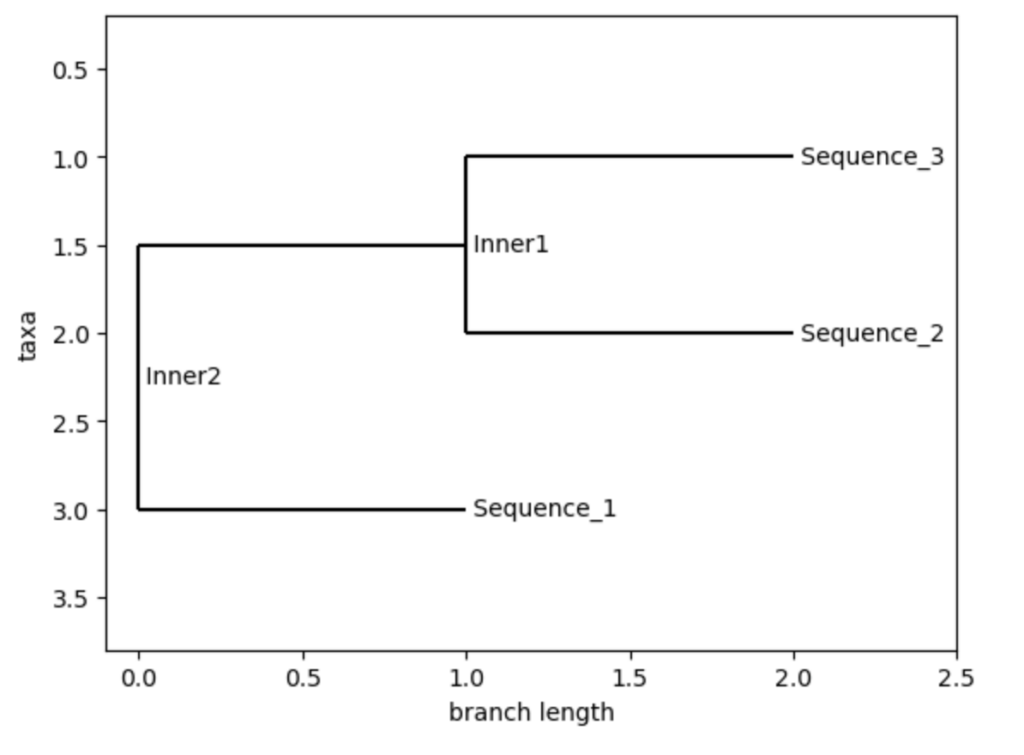
The following example shows how to create a phylogenetic tree in Python. There needs to exist a file with already aligned fasta sequences like this:
>Sequence_1
ATCGGCTAGCTAGCTAGCTAGCTAGCTAGCT
>Sequence_2
ATCGGCTAGCTAGCTAGCTAGCTAGCTAGCT
>Sequence_3
ATCGGCTAGCTAGCTAGCTAGCTAGCTAGCTThen the code to create the tree is:
from Bio import AlignIO
from Bio import Phylo
from Bio.Phylo.TreeConstruction import DistanceCalculator, DistanceTreeConstructor
# Read the already aligned sequences
aligned_file = "sequences.fasta"
alignment = AlignIO.read(aligned_file, "fasta")
# Calculate distance matrix
calculator = DistanceCalculator('identity')
distance_matrix = calculator.get_distance(alignment)
# Build tree using UPGMA method
constructor = DistanceTreeConstructor(calculator, 'upgma')
tree = constructor.build_tree(alignment)
# Draw the tree
Phylo.draw(tree)